
Diels-Alder(D-A)反应是一类能够直接形成C-C键的重要有机反应,可以高效地实现对于杂环、手性螺环和桥环等复杂结构的构筑,被广泛地应用于合成化学领域;近年来一些特殊的D-A反应还被开发为“点击化学”(click chemistry)和“生物正交化学”(bioorthogonal chemistry),并应用于材料科学和生命科学的研究中。通过对于天然产物生源合成途径的分析,诸多天然产物的骨架构筑过程中可能涉及到D-A反应,因此D-A反应也经常被巧妙地应用于天然产物的仿生合成研究中。一方面,随着分子前线轨道理论的发展,合成化学家开发了一系列能够促进D-A反应的小分子催化剂;另一方面,基于著名化学家鲍林的酶催化理论(生物大分子在催化过程中可以通过主-客体相互作用稳定底物小分子的反应过渡态),人工筛选、制备的能够识别底物反应过渡态的抗体酶或核酶也能够在一定程度上催化分子间的D-A反应。然而,由于缺乏直接的结构生物学和生物化学证据,天然产物的生物合成过程中所涉及到的D-A反应是否由酶催化完成以及相应的催化机制目前尚不为人知。近期,中国科学院上海有机化学研究所生命有机化学国家重点实验室刘文研究员课题组与潘李锋研究员课题组合作,在国际上首次报道了源于吡咯吲哚霉素(pyrroindomycin,PYR)生物合成途径中的D-A酶PyrI4及其结合底物小分子复合物的结构,并综合运用生物化学、蛋白质核磁共振等研究方法,系统性地阐明了以该蛋白为代表的酶促D-A反应催化机理,相关成果发表于Cell子刊Cell Chemical Biology杂志上(Cell Chem. Biol., 2016, 23, 352-360.)。
PYR是一类来源于链霉菌(Streptomyces rugosporus)的螺环乙酰乙酸内酰胺类抗生素,其对于耐甲氧西林金黄色葡萄球菌(MRSA)和耐万古霉素的肠球菌(VRE)均有较强的杀伤作用;另外该分子结构中含有一个[6,6]并环和一个[6,5]螺环,这样两个可能由酶促D-A反应构筑的结构单元引起了合成化学家和生物化学家们极大的兴趣。在前期的研究中,刘文课题组在国际上首次报道了PYR的生物合成基因簇(J. Am. Chem. Soc., 2012, 134, 17342-17345.),又通过体内(in vivo)、体外(in vitro)实验相结合,首次阐述了PYR分子母核的刚性五环骨架需要由连续两步的酶促D-A反应构筑(Nat. Chem. Biol., 2015, 11, 259-265.),其中PyrI4催化了用于构筑[6,5]螺环结构的D-A反应。
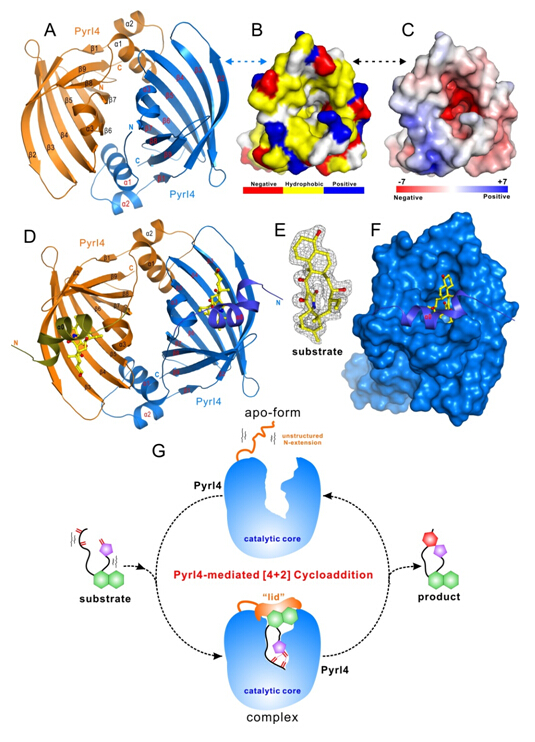
在本研究中,潘李锋课题组与刘文课题组展开合作,他们首先解析了PyrI4蛋白酶的晶体结构(apo-型),发现其具备由β-桶状结构构成的刚性骨架;然后通过大量的条件筛选,他们又获得了PyrI4结合底物小分子的过渡态的复合物晶体结构(holo-型),并基于结构通过进一步的生物化学研究阐明了该蛋白酶的催化活性空腔与重要氨基酸残基;随后,通过比较apo-型与holo-型蛋白结构的差异并结合蛋白核磁共振分析,他们发现PyrI4蛋白N端的前22个氨基酸具有柔性很大的无序结构,而当底物存在时,该部分序列会参与底物分子的识别,并通过与底物以及β-筒状结构之间的特异性相互作用,诱导其自身形成一个特殊的α-螺旋结构,像“盖子”一样把底物分子挤压到β-筒状结构的空腔中,这样“盖状”结构的形成能够释放大量的能量,由此将不利于反应发生的“熵减效应”转化为利于反应进行的“焓效应”。最后,根据一系列的生化实验和理论计算,他们最终提出PyrI4作为催化PYR[6,5]螺环结构形成过程中D-A反应的高效催化剂,主要发挥在如下几个方面作用:①作为“熵陷阱”(entropy trap),有效地将长链底物中二烯体和亲二烯体两部分结构折叠成能够有利于发生D-A反应的构象(即D-A反应的过渡态构象);②活性位点处的Q115氨基酸残基能够通过与底物亲双烯体部分形成双氢键,降低该部分的LUMO能量,从而促进反应的发生;③活性空腔中富含酸性氨基酸,酸性的空腔能够实现质子转移,防止pKa较低的酸性底物带有负电荷;④由底物诱导的N端“盖状”结构能够通过在蛋白残基之间形成大量氢键和盐键而释放能量,从而稳定蛋白-小分子复合物的结构,并将原本对反应不利的熵减效应转化为对反应有利的焓效应。
在此前的研究中,国际上仅有两例[4+2]环加成酶的晶体结构被报道,却都因为缺乏底物-蛋白复合物的结构而未能阐明其催化机制;另外,这两例报道也均存在较大的争议。PyrI4蛋白结构和功能的阐明,证实了自然界中存在天然的类似于人工抗体酶的催化机制:酶与底物结合放出能量后,两者相互的构象诱导使得小分子的构象与D-A反应过渡态的构象极为相似,由此降低了活化能(Ea)并大大促进了反应的速率,而这一切正源于自然界中蛋白质与小分子的共同进化。通过与螺醇缩酮类天然产物生物合成途径的对比,还可将此类“底物过渡态构象折叠”酶的反应机制推广到更多的酶促协同反应中(Curr.Opin. Chem. Biol., 2016, 31, 95-102.)。本研究为今后人工设计、开发协同反应的生物大分子催化剂提供了重要的实验基础和方向,并将在未来的合成化学和合成生物学研究中发挥重要的作用。
刘文课题组的博士生郑庆飞和潘李锋课题组的硕士生郭玉娇为本文的共同第一作者。上述研究工作得到国家青年千人计划项目、科技部973计划青年专题项目、国家自然科学基金委面上项目、生命有机化学国家重点实验室、科技部、上海市科委以及中国科学院的大力资助。
附件下载: