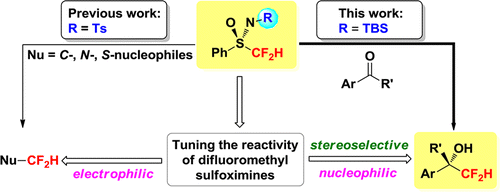
Among various fluoroalkyl groups, the difluoromethyl (CF2H) group is of particular interest, as it is known to be isosteric and isopolar to an OH or SH unit, and can act as a hydrogen donor through hydrogen bonding. Therefore, the difluoromethylated analogues of biologically active compounds are strong candidates for pharmaceuticals. In some cases, difluoromethylated compounds exhibit increased bioactivity compared to their trifluoromethylated counterparts. Over the past decades, a variety of protocols have been developed for introducing the trifluoromethyl group. However, there are few mild and efficient methods for difluoromethylations, and stereoselective difluoromethylation methods of carbonyl compounds are particularly sparse. Recently, we reported the first homochiral α-fluoro sulfoximine-mediated stereoselective fluoroalkylation reaction, and a series of monofluorinated cyclopropanes were synthesized in high yield and with excellent stereoselectivity using (R)-PhSO(NTs)CH2F (Angew. Chem. Int. Ed. 2012, 51, 6966. See Scheme 1). In light of the ability of the sulfoximine group to give high levels of chiral induction, we developeded a method for the stereoselective synthesis of enantiomerically enriched difluoromethyl tertiary alcohols by tuning the reactivity of difluoromethyl sulfoximines from electrophilic (Org. Lett. 2009, 11, 2109.) to nucleophilic (JACS. 2012, accepted) difluoromethylating agents (See Scheme 2). The key feature of this chemistry is the diastereoselective addition of difluoromethyl sulfoximine to the prochiral carbon center of ketones. The present method has also been used to prepare enantiomerically enriched difluoromethyl secondary alcohols as well as two difluorinated analogues of natural products (difluorinated gossonorol 7gand boivinian B 13), which demonstrates the potency of the method.
|
|